
SARM1 Ablation Is Protective and Preserves Spatial Vision in an In Vivo Mouse Model of Retinal Ganglion Cell Degeneration

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
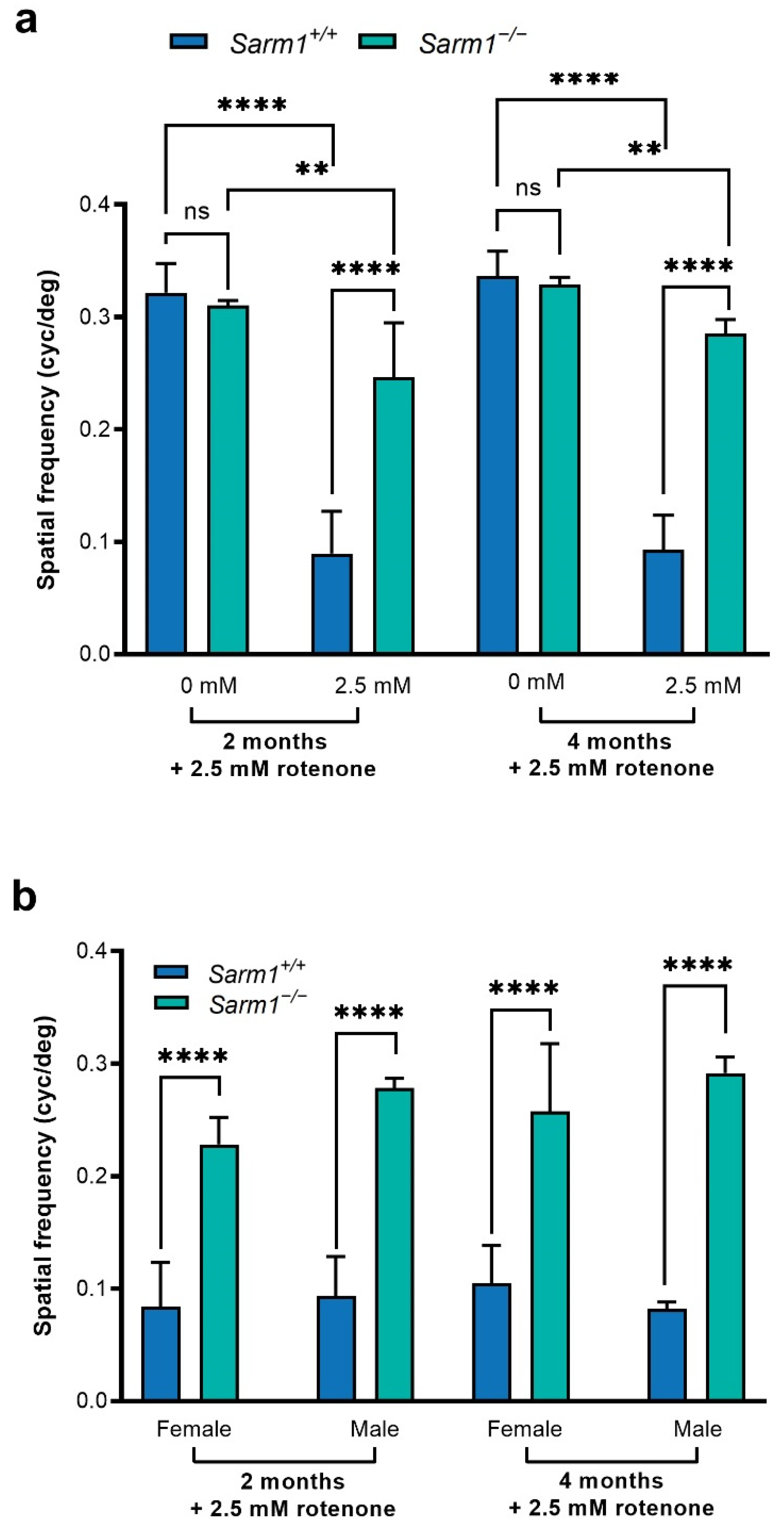
2.1. Protection against Loss of Spatial Vision
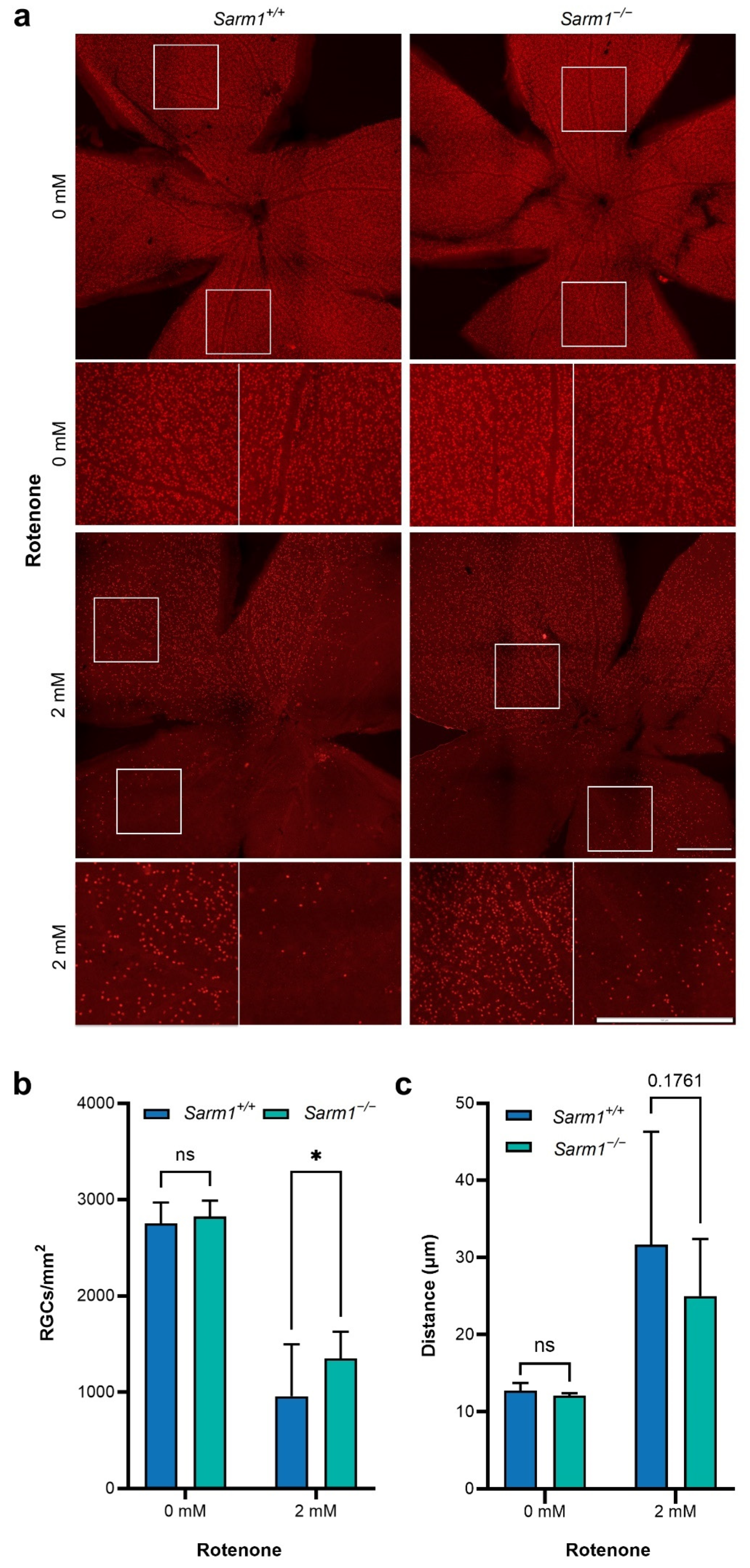
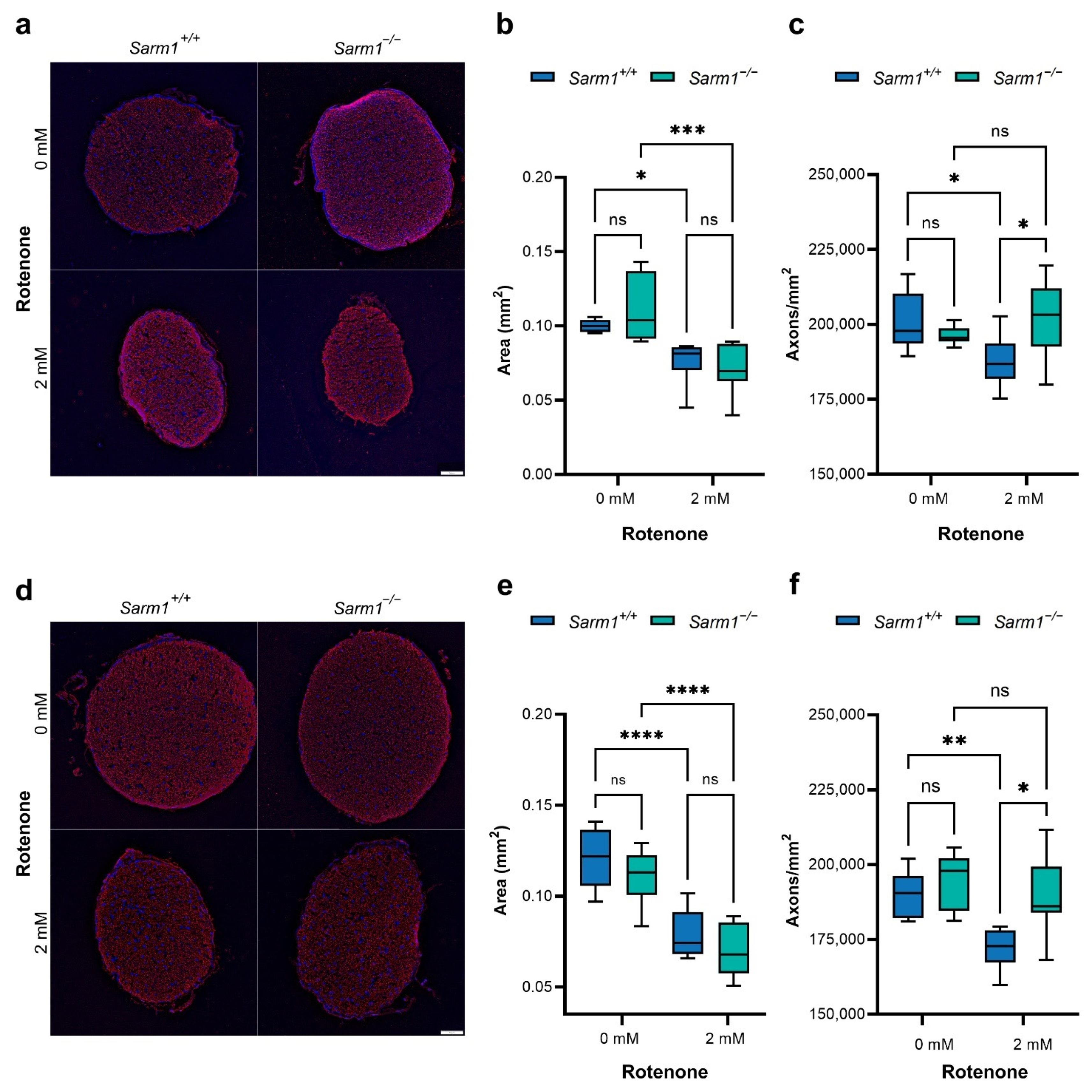
2.2. Protection of RGCs and Their Axons
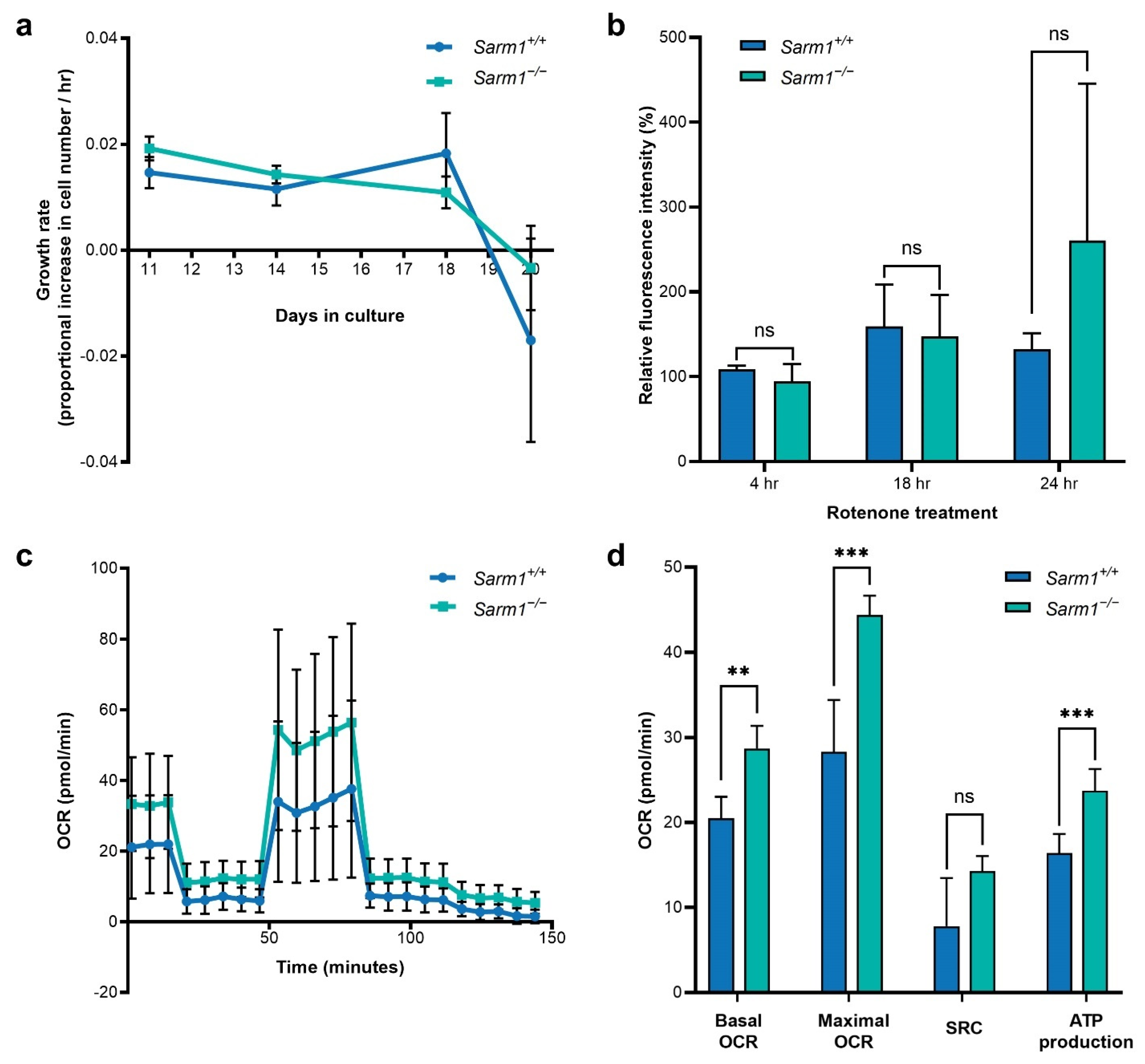
2.3. Bioenergetics of Primary Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Animals and Intravitreal Injections
4.2. Optokinetic Response Measurements
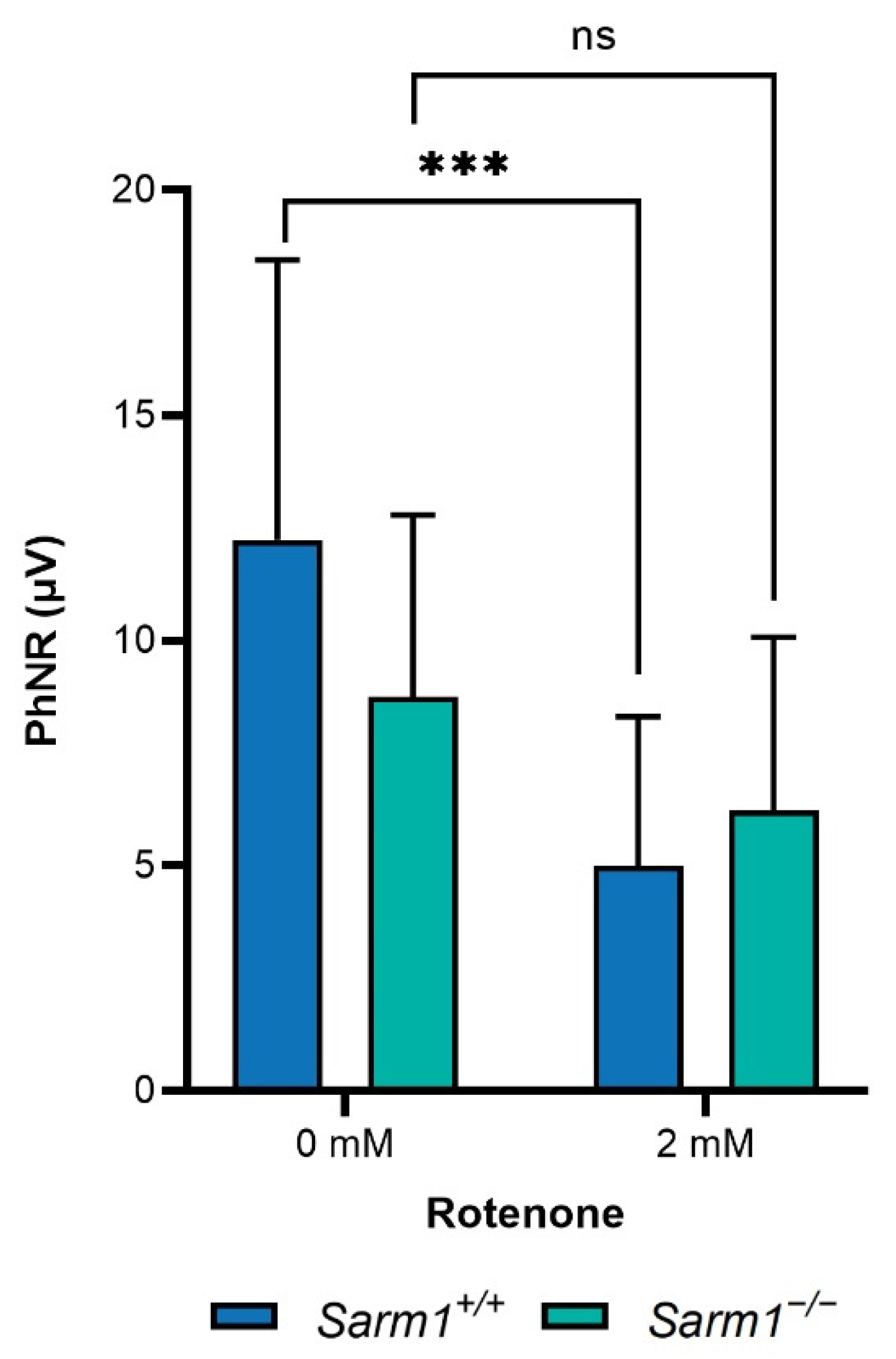
4.3. Photopic Negative Response
4.4. Histology
4.5. Isolation of Primary Fibroblasts from Mouse Tails
4.6. Growth Assays
4.7. Analysis of ROS Accumulation
4.8. Analysis of Mitochondrial Function
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carelli, V.; La Morgia, C.; Ross-Cisneros, F.N.; Sadun, A.A. Optic neuropathies: The tip of the neurodegeneration iceberg. Hum. Mol. Genet. 2017, 26, R139–R150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Morgia, C.; Ross-Cisneros, F.N.; Koronyo, Y.; Hannibal, J.; Gallassi, R.; Cantalupo, G.; Sambati, L.; Pan, B.X.; Tozer, K.R.; Barboni, P.; et al. Melanopsin retinal ganglion cell loss in Alzheimer disease. Ann. Neurol. 2016, 79, 90–109. [Google Scholar] [CrossRef] [PubMed]
- Alqawlaq, S.; Flanagan, J.G.; Sivak, J.M. All roads lead to glaucoma: Induced retinal injury cascades contribute to a common neurodegenerative outcome. Exp. Eye Res. 2019, 183, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Raff, M.C.; Whitmore, A.V.; Finn, J.T. Axonal self-destruction and neurodegeneration. Science 2002, 296, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Libby, R.T.; Li, Y.; Savinova, O.V.; Barter, J.; Smith, R.S.; Nickells, R.W.; John, S.W.M. Susceptibility to neurodegeneration in a glaucoma is modified by bax gene dosage. PLoS Genet. 2005, 1, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syc-Mazurek, S.B.; Fernandes, K.A.; Libby, R.T. JUN is important for ocular hypertension-induced retinal ganglion cell degeneration. Cell Death Dis. 2017, 8, e2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaffarieh, A.; Levin, L.A. Optic nerve disease and axon pathophysiology. In International Review of Neurobiology; Academic Press Inc.: Cambridge, MA, USA, 2012; Volume 105, pp. 1–17. [Google Scholar] [CrossRef]
- Galetta, S.L.; Villoslada, P.; Levin, N.; Shindler, K.; Ishikawa, H.; Parr, E.; Cadavid, D.; Balcer, L.J. Acute optic neuritis. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e135. [Google Scholar] [CrossRef] [Green Version]
- Schlaepfer, W.W. Vincristine-induced axonal alterations in rat peripheral nerve. J. Neuropathol. Exp. Neurol. 1971, 30, 488–505. [Google Scholar] [CrossRef]
- Wang, M.S.; Fang, G.; Culver, D.G.; Davis, A.A.; Rich, M.M.; Glass, J.D. The Wlds protein protects against axonal degeneration: A model of gene therapy for peripheral neuropathy. Ann. Neurol. 2001, 50, 773–779. [Google Scholar] [CrossRef]
- Massoll, C.; Mando, W.; Chintala, S.K. Excitotoxicity Upregulates SARM1 Protein Expression and Promotes Wallerian-Like Degeneration of Retinal Ganglion Cells and Their Axons. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2771. [Google Scholar] [CrossRef]
- Finn, J.T.; Weil, M.; Archer, F.; Siman, R.; Srinivasan, A.; Raff, M.C. Evidence that wallerian degeneration arid localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. J. Neurosci. 2000, 20, 1333–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syc-Mazurek, S.B.; Libby, R.T. Axon injury signaling and compartmentalized injury response in glaucoma. Prog. Retin. Eye Res. 2019, 73, 100769. [Google Scholar] [CrossRef] [PubMed]
- Summers, D.W.; Diantonio, A.; Milbrandt, J. Mitochondrial Dysfunction Induces Sarm1-Dependent Cell Death in Sensory Neurons. J. Neurosci. 2014, 34, 9338–9350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerdts, J.; Summers, D.W.; Sasaki, Y.; DiAntonio, A.; Milbrandt, J. Sarm1-Mediated Axon Degeneration Requires Both SAM and TIR Interactions. J. Neurosci. 2013, 33, 13569–13580. [Google Scholar] [CrossRef]
- Lunn, E.R.; Perry, V.H.; Brown, M.C.; Rosen, H.; Gordon, S. Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur. J. Neurosci. 1989, 1, 27–33. [Google Scholar] [CrossRef]
- Figley, M.D.; DiAntonio, A. The SARM1 axon degeneration pathway: Control of the NAD+ metabolome regulates axon survival in health and disease. Curr. Opin. Neurobiol. 2020, 63, 59–66. [Google Scholar] [CrossRef]
- Conforti, L.; Gilley, J.; Coleman, M.P. Wallerian degeneration: An emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 2014, 15, 394–409. [Google Scholar] [CrossRef]
- Osterloh, J.M.; Yang, J.; Rooney, T.M.; Fox, A.N.; Adalbert, R.; Powell, E.H.; Sheehan, A.E.; Avery, M.A.; Hackett, R.; Logan, M.A.; et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 2012, 337, 481–484. [Google Scholar] [CrossRef] [Green Version]
- Carty, M.; Bowie, A.G. SARM: From immune regulator to cell executioner. Biochem. Pharmacol. 2019, 161, 52–62. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef]
- Szretter, K.J.; Samuel, M.A.; Gilfillan, S.; Fuchs, A.; Colonna, M.; Diamond, M.S. The Immune Adaptor Molecule SARM Modulates Tumor Necrosis Factor Alpha Production and Microglia Activation in the Brainstem and Restricts West Nile Virus Pathogenesis. J. Virol. 2009, 83, 9329–9338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carty, M.; Kearney, J.; Shanahan, K.A.; Lavelle, E.C.; Fallon, P.G.; Bowie, A.G. Cell Survival and Cytokine Release after Inflammasome Activation Is Regulated by the Toll-IL-1R Protein SARM. Immunity 2019, 50, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Essuman, K.; Summers, D.W.; Sasaki, Y.; Mao, X.; DiAntonio, A.; Milbrandt, J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD+ Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 2017, 93, 1334–1343.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, S.; Huang, S.X.; Strickland, A.; Doan, R.A.; Summers, D.W.; Mao, X.; Park, J.; DiAntonio, A.; Milbrandt, J. Gene therapy targeting SARM1 blocks pathological axon degeneration in mice. J. Exp. Med. 2019, 216, 294–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horsefield, S.; Burdett, H.; Zhang, X.; Manik, M.K.; Shi, Y.; Chen, J.; Qi, T.; Gilley, J.; Lai, J.-S.; Rank, M.X.; et al. NAD+ cleavage activity by animal and plant TIR domains in cell death pathways. Science 2019, 365, 793–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, P.A.; Harder, J.M.; John, S.W.M. Glaucoma as a Metabolic Optic Neuropathy: Making the Case for Nicotinamide Treatment in Glaucoma. J. Glaucoma 2017, 26, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Mack, T.G.A.; Reiner, M.; Beirowski, B.; Mi, W.; Emanuelli, M.; Wagner, D.; Thomson, D.; Gillingwater, T.; Court, F.; Conforti, L.; et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat. Neurosci. 2001, 4, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Gerdts, J.; Brace, E.J.; Sasaki, Y.; DiAntonio, A.; Milbrandt, J. SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 2015, 348, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figley, M.D.; Gu, W.; Nanson, J.D.; Shi, Y.; Sasaki, Y.; Cunnea, K.; Malde, A.K.; Jia, X.; Luo, Z.; Saikot, F.K.; et al. SARM1 is a metabolic sensor activated by an increased NMN/NAD+ ratio to trigger axon degeneration. Neuron 2021, 109, 1118–1136.e11. [Google Scholar] [CrossRef]
- Bratkowski, M.; Xie, T.; Thayer, D.A.; Lad, S.; Mathur, P.; Yang, Y.S.; Danko, G.; Burdett, T.C.; Danao, J.; Cantor, A.; et al. Structural and Mechanistic Regulation of the Pro-degenerative NAD Hydrolase SARM1. Cell Rep. 2020, 32, 107999. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, T.; Lee, C.-H.; Chang, Q.; Yang, J.; Zhang, Z. The NAD+-mediated self-inhibition mechanism of pro-neurodegenerative SARM1. Nature 2020, 588, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, E.; Gibbons, L.; Neto, N.G.; Kenna, P.; Carty, M.; Humphries, M.; Humphries, P.; Campbell, M.; Monaghan, M.; Bowie, A.; et al. SARM1 deficiency promotes rod and cone photoreceptor cell survival in a model of retinal degeneration. Life Sci. Alliance 2020, 3, e201900618. [Google Scholar] [CrossRef] [PubMed]
- Turkiew, E.; Falconer, D.; Reed, N.; Höke, A. Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. J. Peripher. Nerv. Syst. 2017, 22, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Liu, J.; Luan, Y.; Liu, Z.; Lai, H.; Zhong, W.; Yang, Y.; Yu, H.; Feng, N.; Wang, H.; et al. Sarm1 Gene Deficiency Attenuates Diabetic Peripheral Neuropathy in Mice. Diabetes 2019, 68, 2120–2130. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wu, Z.; Renier, N.; Simon, D.J.; Uryu, K.; Park, D.S.; Greer, P.A.; Tournier, C.; Davis, R.J.; Tessier-Lavigne, M. Pathological axonal death through a Mapk cascade that triggers a local energy deficit. Cell 2015, 160, 161–176. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-H.; Sasaki, Y.; DiAntonio, A.; Milbrandt, J. SARM1 is required in human derived sensory neurons for injury-induced and neurotoxic axon degeneration. Exp. Neurol. 2021, 339, 113636. [Google Scholar] [CrossRef]
- Van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; Van Der Spek, R.A.A.; Võsa, U.; De Jong, S.; Robinson, M.R.; et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogh, I.; Ratti, A.; Gellera, C.; Lin, K.; Tilcoa, C.; Moskvina, V.; Corrado, L.; Sorarù, G.; Cereda, C.; Corti, S.; et al. A genome-wide association meta-analysis identifies a novel locus at 17q11. 2 associated with sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 2014, 23, 2220–2231. [Google Scholar] [CrossRef]
- Murata, H.; Cho Khine, C.; Nishikawa, A.; Yamamoto, K.-I.; Kinoshita, R.; Sakaguchi, M. c-Jun N-terminal kinase (JNK)-mediated phosphorylation of SARM1 regulates NAD cleavage activity to inhibit mitochondrial respiration. J. Biol. Chem. 2018, 293, 18933–18943. [Google Scholar] [CrossRef]
- Loreto, A.; Angeletti, C.; Gilley, J.; Arthur-Farraj, P.; Merlini, E.; Amici, A.; Desrochers, L.M.; Wang, Q.; Orsomando, G.; Coleman, M.P.; et al. Potent activation of SARM1 by NMN analogue VMN underlies vacor neurotoxicity. Biorxiv Neurosci. 2021, 2020, 18.304261. [Google Scholar] [CrossRef]
- Summers, D.W.; Frey, E.; Walker, L.J.; Milbrandt, J.; DiAntonio, A. DLK Activation Synergizes with Mitochondrial Dysfunction to Downregulate Axon Survival Factors and Promote SARM1-Dependent Axon Degeneration. Mol. Neurobiol. 2020, 57, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Tapias, V.; Na, H.M.; Honick, A.S.; Drolet, R.E.; Greenamyre, J.T. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis. 2009, 34, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadderton, N.; Palfi, A.; Millington-Ward, S.; Gobbo, O.; Overlack, N.; Carrigan, M.; O’Reilly, M.; Campbell, M.; Ehrhardt, C.; Wolfrum, U.; et al. Intravitreal delivery of AAV-NDI1 provides functional benefit in a murine model of Leber hereditary optic neuropathy. Eur. J. Hum. Genet. 2013, 21, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jones, D.; Gonzalez-Lima, F. Mouse model of optic neuropathy caused by mitochondrial complex I dysfunction. Neurosci. Lett. 2002, 326, 97–100. [Google Scholar] [CrossRef]
- Mansergh, F.C.; Chadderton, N.; Kenna, P.F.; Gobbo, O.L.; Farrar, G.J. Cell therapy using retinal progenitor cells shows therapeutic effect in a chemically-induced rotenone mouse model of Leber hereditary optic neuropathy. Eur. J. Hum. Genet. 2014, 22, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Degli Esposti, M. Inhibitors of NADH-ubiquinone reductase: An overview. Biochim. Biophys. Acta Bioenerg. 1998, 1364, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochim. Biophys. Acta 2009, 1787, 384–392. [Google Scholar] [CrossRef]
- Heinz, S.; Freyberger, A.; Lawrenz, B.; Schladt, L.; Schmuck, G.; Ellinger-Ziegelbauer, H. Mechanistic Investigations of the Mitochondrial Complex i Inhibitor Rotenone in the Context of Pharmacological and Safety Evaluation. Sci. Rep. 2017, 7, 45465. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y.; Engber, T.M.; Hughes, R.O.; Figley, M.D.; Wu, T.; Bosanac, T.; Devraj, R.; Milbrandt, J.; Krauss, R.; DiAntonio, A. cADPR is a gene dosage-sensitive biomarker of SARM1 activity in healthy, compromised, and degenerating axons. Exp. Neurol. 2020, 329, 113252. [Google Scholar] [CrossRef]
- Hughes, R.O.; Bosanac, T.; Mao, X.; Engber, T.M.; DiAntonio, A.; Milbrandt, J.; Devraj, R.; Krauss, R. Small Molecule SARM1 Inhibitors Recapitulate the SARM1−/− Phenotype and Allow Recovery of a Metastable Pool of Axons Fated to Degenerate. Cell Rep. 2021, 34, 108588. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Griffiths, P.G.; Chinnery, P.F. Mitochondrial optic neuropathies-Disease mechanisms and therapeutic strategies. Prog. Retin. Eye Res. 2011, 30, 81–114. [Google Scholar] [CrossRef] [Green Version]
- Zetterberg, M. Age-related eye disease and gender. Maturitas 2016, 83, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Tenkorang, M.A.; Snyder, B.; Cunningham, R.L. Sex-related differences in oxidative stress and neurodegeneration. Steroids 2018, 133, 21–27. [Google Scholar] [CrossRef] [PubMed]
- EUGenMed; Cardiovascular Clinical Study Group; Regitz-Zagrosek, V.; Oertelt-Prigione, S.; Prescott, E.; Franconi, F.; Gerdts, E.; Foryst-Ludwig, A.; Maas, A.H.E.M.; Kautzky-Willer, A.; et al. Gender in cardiovascular diseases: Impact on clinical manifestations, management, and outcomes. Eur. Heart J. 2015, 37, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Ngo, S.T.; Steyn, F.J.; McCombe, P.A. Gender differences in autoimmune disease. Front. Neuroendocrinol. 2014, 35, 347–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brocher, J. The BioVoxxel image processing and analysis toolbox. In Proceedings of the EuBIAS Conference, Paris, France, 5 January 2015; Institut Curie: Paris, France, 2015. [Google Scholar]
- McDougald, D.S.; Dine, K.E.; Zezulin, A.U.; Bennett, J.; Shindler, K.S. SIRT1 and NRF2 Gene Transfer Mediate Distinct Neuroprotective Effects Upon Retinal Ganglion Cell Survival and Function in Experimental Optic Neuritis. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1212–1220. [Google Scholar] [CrossRef] [Green Version]
- Beck, R.W.; Cleary, P.A.; Backlund, J.; Yu, C. The Course of Visual Recovery after Optic Neuritis: Experience of the Optic Neuritis Treatment Trial. Ophthalmology 1994, 101, 1771–1778. [Google Scholar] [CrossRef]
- Ko, K.W.; Milbrandt, J.; DiAntonio, A. SARM1 acts downstream of neuroinflammatory and necroptotic signaling to induce axon degeneration. J. Cell Biol. 2020, 219, e201912047. [Google Scholar] [CrossRef]
- White, M.A.; Lin, Z.; Kim, E.; Henstridge, C.M.; Pena Altamira, E.; Hunt, C.K.; Burchill, E.; Callaghan, I.; Loreto, A.; Brown-Wright, H.; et al. Sarm1 deletion suppresses TDP-43-linked motor neuron degeneration and cortical spine loss. Acta Neuropathol. Commun. 2019, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, K.A.; Mitchell, K.L.; Patel, A.; Marola, O.J.; Shrager, P.; Zack, D.J.; Libby, R.T.; Welsbie, D.S. Role of SARM1 and DR6 in retinal ganglion cell axonal and somal degeneration following axonal injury. Exp. Eye Res. 2018, 171, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Prusky, G.T.; Alam, N.M.; Beekman, S.; Douglas, R.M. Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Investig. Opthalmology Vis. Sci. 2004, 45, 4611–4616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, R.M.; Alam, N.M.; Silver, B.D.; Mcgill, T.J.; Tschetter, W.W.; Prusky, G.T. Independent visual threshold measurements in the two eyes of freely moving rats and mice using a virtual-reality optokinetic system. Vis. Neurosci. 2005, 22, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Pinares-Garcia, P.; Stratikopoulos, M.; Zagato, A.; Loke, H.; Lee, J. Sex: A significant risk factor for neurodevelopmental and neurodegenerative disorders. Brain Sci. 2018, 8, 154. [Google Scholar] [CrossRef] [Green Version]
- Ffrench-Constant, C.; Miller, R.H.; Burne, J.F.; Raff, M.C. Evidence that migratory oligodendrocyte-type-2 astrocyte (O-2A) progenitor cells are kept out of the rat retina by a barrier at the eye-end of the optic nerve. J. Neurocytol. 1988, 17, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weimer, R.M.; Kallop, D.; Olsen, O.; Wu, Z.; Renier, N.; Uryu, K.; Tessier-Lavigne, M. Regulation of axon degeneration after injury and in development by the endogenous calpain inhibitor calpastatin. Neuron 2013, 80, 1175–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu-Wai-Man, P.; Soiferman, D.; Moore, D.G.; Burté, F.; Saada, A. Evaluating the therapeutic potential of idebenone and related quinone analogues in Leber hereditary optic neuropathy. Mitochondrion 2017, 36, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Antony, P.M.A.; Kondratyeva, O.; Mommaerts, K.; Ostaszewski, M.; Sokolowska, K.; Baumuratov, A.S.; Longhino, L.; Poulain, J.F.; Grossmann, D.; Balling, R.; et al. Fibroblast mitochondria in idiopathic Parkinson’s disease display morphological changes and enhanced resistance to depolarization. Sci. Rep. 2020, 10, 1569. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Vohra, B.P.S.; Lund, F.E.; Milbrandt, J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J. Neurosci. 2009, 29, 5525–5535. [Google Scholar] [CrossRef]
- Farrar, G.J.; Chadderton, N.; Kenna, P.F.; Millington-Ward, S. Mitochondrial disorders: Aetiologies, models systems, and candidate therapies. Trends Genet. 2013, 29, 488–497. [Google Scholar] [CrossRef] [Green Version]
- Monzio Compagnoni, G.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The Role of Mitochondria in Neurodegenerative Diseases: The Lesson from Alzheimer’s Disease and Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 2959–2980. [Google Scholar] [CrossRef]
- Schapira, A.H.V. Complex I: Inhibitors, inhibition and neurodegeneration. Exp. Neurol. 2010, 224, 331–335. [Google Scholar] [CrossRef] [PubMed]
- SanGiovanni, J.P.; Arking, D.E.; Iyengar, S.K.; Elashoff, M.; Clemons, T.E.; Reed, G.F.; Henning, A.K.; Sivakumaran, T.A.; Xu, X.; DeWan, A.; et al. Mitochondrial DNA variants of respiratory complex I that uniquely characterize haplogroup T2 are associated with increased risk of age-related macular degeneration. PLoS ONE 2009, 4, e5508. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Sheck, L.; Crowston, J.G.; van Bergen, N.J.; O’Neill, E.C.; O’Hare, F.; Kong, Y.X.G.; Chrysostomou, V.; Vincent, A.L.; Trounce, I.A. Impaired complex-I-Linked respiration and ATP synthesis in primary open-angle glaucoma patient lymphoblasts. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2431–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palfi, A.; Hokamp, K.; Hauck, S.M.; Vencken, S.; Millington-Ward, S.; Chadderton, N.; Carrigan, M.; Kortvely, E.; Greene, C.M.; Kenna, P.F.; et al. MicroRNA regulatory circuits in a mouse model of inherited retinal degeneration. Sci. Rep. 2016, 6, 31431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palfi, A.; Yesmambetov, A.; Humphries, P.; Hokamp, K.; Farrar, G.J. Non-photoreceptor Expression of Tulp1 May Contribute to Extensive Retinal Degeneration in Tulp1−/− Mice. Front. Neurosci. 2020, 14, 656. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Gasser, S. Generating primary fibroblast cultures from mouse ear and tail tissues. J. Vis. Exp. 2016, 107, e53565. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Finnegan, L.K.; Chadderton, N.; Kenna, P.F.; Palfi, A.; Carty, M.; Bowie, A.G.; Millington-Ward, S.; Farrar, G.J. SARM1 Ablation Is Protective and Preserves Spatial Vision in an In Vivo Mouse Model of Retinal Ganglion Cell Degeneration. Int. J. Mol. Sci. 2022, 23, 1606. https://doi.org/10.3390/ijms23031606
Finnegan LK, Chadderton N, Kenna PF, Palfi A, Carty M, Bowie AG, Millington-Ward S, Farrar GJ. SARM1 Ablation Is Protective and Preserves Spatial Vision in an In Vivo Mouse Model of Retinal Ganglion Cell Degeneration. International Journal of Molecular Sciences. 2022; 23(3):1606. https://doi.org/10.3390/ijms23031606
Chicago/Turabian StyleFinnegan, Laura K., Naomi Chadderton, Paul F. Kenna, Arpad Palfi, Michael Carty, Andrew G. Bowie, Sophia Millington-Ward, and G. Jane Farrar. 2022. "SARM1 Ablation Is Protective and Preserves Spatial Vision in an In Vivo Mouse Model of Retinal Ganglion Cell Degeneration" International Journal of Molecular Sciences 23, no. 3: 1606. https://doi.org/10.3390/ijms23031606